



The FDA recently issued a discussion paper and request for feedback titled, “Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning [AI/ML]-Based Software as a Medical Device.” The challenge artificial intelligence (AI) and machine learning (ML) present to FDA device regulation is straightforward: the FDA must approve or clear devices as safe and effective, yet some forms of AI or ML software can adapt and change as they obtain new data or otherwise gain experience.
The FDA has requested comments by June 3, 2019. Given the complexity of the issues, it would not be surprising to see the FDA extend this date.
One of the main questions is: How can the FDA determine a product is safe and effective when the product by its nature is expected to change after the determination is made?
Indeed, the discussion paper concedes that “the traditional paradigm of medical device regulation was not designed for adaptive AI/ML technologies.” In the paper, the FDA proposes to address this challenge by adopting “a new, total product lifecycle (TPLC) regulatory approach that facilitates a rapid cycle of product improvement and allows these devices to continually improve while providing effective safeguards.”
The FDA’s proposed approach is summarized in the following graphic that is found in the discussion paper:
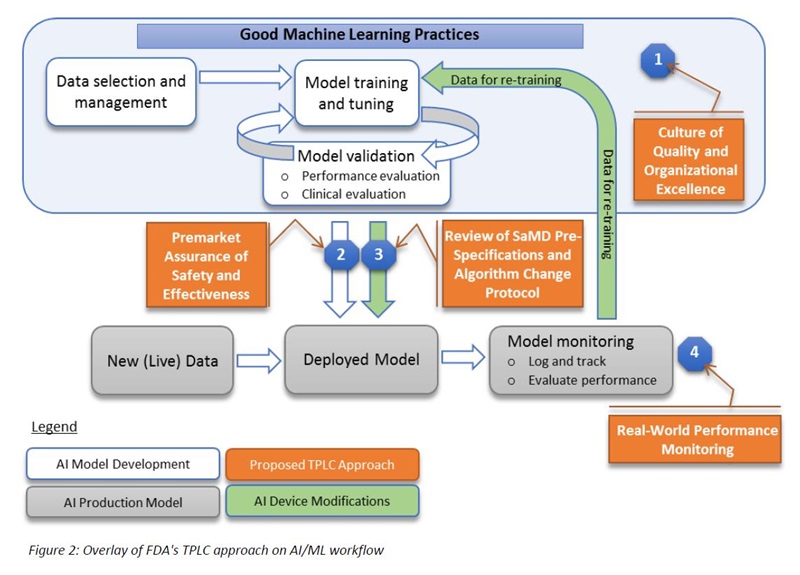
Describing this graphic, the paper states:
“… the FDA’s proposed TPLC approach is based on the following general principles that balance the benefits and risks, and provide access to safe and effective AI/ML-based SaMD (Software as a Medical Device):
1. Establish clear expectations on quality systems and good ML practices (GMLP);
2. Conduct premarket review for those SaMD that require premarket submission to demonstrate reasonable assurance of safety and effectiveness and establish clear expectations for manufacturers of AI/ML-based SaMD to continually manage patient risks throughout the lifecycle;
3. Expect manufacturers to monitor the AI/ML device and incorporate a risk management approach and other approaches outlined in the “Deciding When to Submit a 510(k) for a Software Change to an Existing Device” Guidance in development, validation, and execution of the algorithm changes; and
4. Enable increased transparency to users and FDA using postmarket real-world performance reporting for maintaining continued assurance of safety and effectiveness.”
The paper is careful to explain that it is just a baby-step in the direction of formulating a regulatory framework for AI/ML-based SaMD: “This discussion paper describes an innovative approach that may require additional statutory authority to implement fully. The proposed framework is being issued for discussion purposes only and is not a draft guidance. This document is … instead meant to seek early input from groups and individuals outside the Agency prior to development of a draft guidance.” Throughout the paper, the FDA sprinkled specific questions on which it seeks outside input.
For more information, please contact the Barnes & Thornburg attorney with whom you work or Lynn Tyler, chair of the firm’s Food, Drug and Device group, at 317-231-7392 or lynn.tyler@btlaw.com.
© 2019 Barnes & Thornburg LLP. All Rights Reserved. This page, and all information on it, is proprietary and the property of Barnes & Thornburg LLP. It may not be reproduced, in any form, without the express written consent of Barnes & Thornburg LLP.
This Barnes & Thornburg LLP publication should not be construed as legal advice or legal opinion on any specific facts or circumstances. The contents are intended for general informational purposes only, and you are urged to consult your own lawyer on any specific legal questions you may have concerning your situation.





/Passle/6488d4630e7e25c9ac9f834a/SearchServiceImages/2024-11-14-13-11-27-495-6735f6fff42d6cc59c8ec5c1.jpg)
/Passle/6488d4630e7e25c9ac9f834a/SearchServiceImages/2024-11-11-22-02-38-042-67327efe31216b909e6ea644.jpg)
